|
Potency and Stability Testing
Potency tests are designed to determine how much of an active drug is in a sample. Stability tests are used to determine an expiration date of a product or a beyond-use date (BUD) of a preparation. Being able to employ the proper method to determine potency or stability is the key to understanding the difference between potency testing versus stability testing. In order to determine potency, a method may or may not be stability indicating. When determining stability, the method must be stability indicating. When using a stability-indicating method, both potency and stability can be determined. Quality-assurance programs are essential to establishing standards for compounded preparations. It is important that compounding pharmacists understand the differences between potency and stability tests and that these tests are made an integral part of the quality-assurance program.
Introduction
Oftentimes the question is asked, "What is the difference between potency and stability?" This seems like a rather simple question and, in some respects, it is. However, to answer this question, one must understand the methods used to analyze the potency and stability of a compound. The most common flaw in determining stability is failure to use an analytical method that has been demonstrated to be stability indicating. So, it is not a surprise that the most important aspect of determining potency and stability are the methods employed in the process. Simply put, a stability-indicating method must be used in order to determine stability. A stability-indicating method has the capabilities of determining potency as well. Potency tests cannot be used to determine stability, unless a stability-indicating method is used. The purpose of this paper is to explain the difference between potency and stability, why they are of importance, and how they are determined. The method used to determine the concentration of the active ingredient is the most critical step in the process.
Potency
Potency can be described as the concentration of the drug in a compounded preparation. Potency tests are known as quantitative tests and are designed to determine how much of a drug is in a sample. High-performance liquid chromatography (HPLC) is the typical methodology employed in determining potency. HPLC is a preferred method because it is very specific and efficient. Although HPLC can be used in stability-indicating methods, not all HPLC procedures are stability indicating, and they must not be assumed to be so. Other methods used to test potency include titration, which uses the principles of chemistry; and microbial assays, which are sometimes used to test antibiotics. Titration is based upon a known chemical reaction with the desired drug. A microbial assay is performed using bacteria and the antibiotic of choice and examining "zones of inhibition." When used alone (without chromatography), ultraviolet-visible spectrophotometry (UV-VIS) can also be employed to determine potency for single analytes only in solutions. Multiple compounds could interfere with ultraviolet (UV) absorption, resulting in erroneous results when using UV-VIS spectrophotometry alone. When performing a potency test, the methods used determine whether or not one will be able to determine stability as well. As mentioned above, a stability-indicating method must be used in order to determine stability.
The purpose of strength, or potency, testing is to establish or verify the concentration (potency) of the drug in the compounded preparation. The United States Pharmacopeia (USP) has established that the acceptable range of most compounded preparations is typically ± 10%, or within the range of 90.0% to 110.0%.
The problem is that many "potency" tests do not separate the intact drug from the degradation products, and they show up under one peak in the chromatogram, giving false information that the drug concentration has not changed—when it actually has. Properly done, a stability-indicating assay will separate out the degradation products/peaks and show the intact drug peak as it decreases in area or height, reflecting a change in the concentration of the intact drug.
Stability
Stability is the extent to which a product retains, within specified limits, and throughout its period of storage and use, the same properties and characteristics that it possessed at the time of its manufacture. The United States Pharmacopeia 35-National Formulary 30 (USP 35-NF 30) provides definitions for five general types of stability:
- Chemical - Each active ingredient retains its chemical integrity and labeled potency, within the specified limits.
- Physical - The original physical properties, including appearance, palatability, uniformity, dissolution, and suspendability, are retained.
- Microbiological - Sterility or resistance to microbial growth is retained according to the specified requirements. Antimicrobial agents that are present retain effectiveness within the specified limits.
- Therapeutic - The therapeutic effect remains unchanged.
- Toxicological - No significant increase in toxicity occurs.
Instability describes chemical reactions that are "…incessant, irreversible, and result in distinctly different chemical entities (degradation products) that can be both therapeutically inactive and possibly exhibit greater toxicity."
Incompatibility is different from instability but must be considered in the overall stability evaluation of a preparation. Incompatibility generally refers to visually evident and "…physicochemical phenomena such as concentration-dependent precipitation and acid-base reactions, with the products of reaction manifested as a change in physical state, including protonation-deprotonation equilibria."
Example
As an example of where some compounding pharmacists run into problems, let's say your pharmacy contracted with an analytical laboratory to run a potency test that did not use stability-indicating methods on your compound and you wanted results at time zero, 30 days, and 60 days. The target concentration of your compound was intended to be 10 mg/mL. The test that will be performed will indicate only potency, not stability. The obvious reason is because the test did not use stability-indicating methods. In other words, at those predefined time points of day 0, 30 days, and 60 days, the lab was only analyzing how much of the compound was present. The lab could not, however, differentiate the compound of interest from degradants or excipients in the preparation that may be "coeluting" in the chromatogram. The results might be reported that the compounded preparation was at a concentration of 10 mg/mL at each time point.
The results cannot be extrapolated to determine a stability of 60 days because in the analysis there could have been degradants or excipients that were present but not detected (again assuming that a stability-indicating method was not used in the analysis). To put it into numbers, the actual concentration of the active ingredient could have been 6 mg/mL with 3 mg/mL of degradants and 1 mg/mL of excipients. The most important thing to realize in this scenario is we can determine potency but not stability because stability-indicating methods were not used. Had stability-indicating methods been used to determine potency, then the results could have been used to determine a BUD, otherwise referred to as stability. Using the previous example, if the concentration at time 60 days was 10 mg/mL and stability-indicating methods were used, we could be sure that we were looking at just the active ingredient.
Stability Testing
Stability testing usually includes method development, method validation, and a stability study. The method development will separate the active ingredient from its degradants and impurities, as well as any other excipients in the preparation. This is done by force degrading the active ingredient and inactive ingredients to ensure that there aren't any degradants interfering with the analysis. In the process of force degradation, high heat and humidity, UV radiation, acid exposure, base exposure, and peroxide exposure are performed on the compound. It is this step that is different from a simple potency test. Figure 1 is an example chromatogram of a stability-indicating HPLC method containing analyte and degradant peaks that are fully resolved from one another. When looking at this chromatogram, it is important to notice that the active ingredient, or analyte, is completely separated from its degradants. Stability can be determined from this type of study simply because stability-indicating methods were used in the analysis. The method validation confirms that the method meets certain criteria. The typical analytical characteristics used in method validation include accuracy, precision, specificity, detection limit, quantitation limit, linearity, range, and ruggedness as outlined in Chapter <1163> in the USP. The stability study includes storing the preparation in stability chambers, testing it at predetermined time points, and then determining stability. These time points can be specified by the compounder or may be dictated by the particular compound. Once again, it is crucial to understand that the methods used to determine stability must be stability indicating. Equally important to understand is that a potency test that uses stability-indicating methods can determine potency as well as stability.
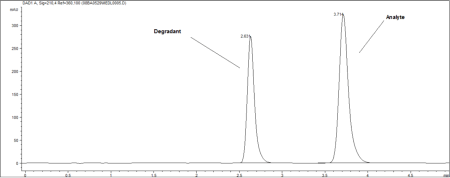
Figure 1. Example chromatogram of a stability-indicating, high-performance liquid chromatography method containing analyte and degradant peaks that are fully resolved from one another.
High-performance Liquid Chromatographic Diode Array Detectors
The analytical method used for the analysis must be "stability indicating," meaning that it can accurately measure the concentration of the active pharmaceutical ingredient (API) in the presence of other chemicals that may be in the pharmaceutical. These interfering chemicals can be either breakdown products or other excipients. With HPLC analysis, the only difficulty comes when an interfering substance elutes from the chromatograph column at the same time as the active API and is hidden under the chromatographic peak of the API. On older HPLC systems, this was a real problem because they only used one UV wavelength in the analysis of the eluting peaks.
The photo diode array (PDA) detector is a device that scans from about 200 nm up to 400 nm in the UV range (and can actually go up to 700 nm in the visible range in some instruments). It takes the full array scan every second or so of the eluent coming from the HPLC. The software starts at the beginning of a peak and scans the eluent (basically by "slicing" it into pieces and doing a complete scan instantly). Then, the scans are compared (overlaid) and any change is identified, and, with the use of an algorithm, the "peak purity" is calculated by comparing the middle peak scans with those as they go out to the leading and trailing tails. If the scans overlay perfectly, then the peak purity will be 100%. If they do not, then it is a calculated percentage. The problem is that a UV scan is not specific and small changes in a drug molecule can occur that may not be picked up in the scan but may alter the drug potency, even though it still looks like it is good. The molecule contains "chromophores" that absorb the UV light at different wavelengths and efficiencies. If a molecule degrades but the change is not in a strong chromophore, then it will not show up and the potency will not be accurately determined.
Peak-purity evaluation should be performed during validation as part of the specificity test of the forced stress samples. The peak-purity test helps to ensure that the method can separate out degradation products during the stability study, and "potency" of the API can be assessed versus the reference standard. One can apply peak-purity analysis to compounded preparations for routine potency testing, and, maybe, time-point testing as part of their BUD. But the method itself still needs to be validated to become a standard monograph method. The PDA method for peak-purity determination can be used to "supplement or support" a stability-indicating analytical method, but should NOT be used in place of it.
Summary
In summary, it is important for the pharmacist who extemporaneously compounds to ensure the strength, quality, identity, and purity of compounded preparations. An outside analytical laboratory can assist by providing quality control and quality assurance. Determination of potency or concentration is invaluable in maintaining a good preparation that is accurate and precise. Remember, to determine the BUD, a stability-indicating HPLC method must be used.
Loyd V. Allen, Jr., PhD, RPh
Editor-in-Chief
International Journal of Pharmaceutical Compounding
Remington: The Science and Practice of Pharmacy
Note: Appreciation is extended to Analytical Research Laboratories (Drs. Tom Kupiec and Nicole Vu) for their contributions and input into this Newsletter. Also, see Kupiec TC, Skinner R, Lanier L. Stability versus potency testing: The madness is in the method. IJPC 2008; 12(1): 50-53.
|